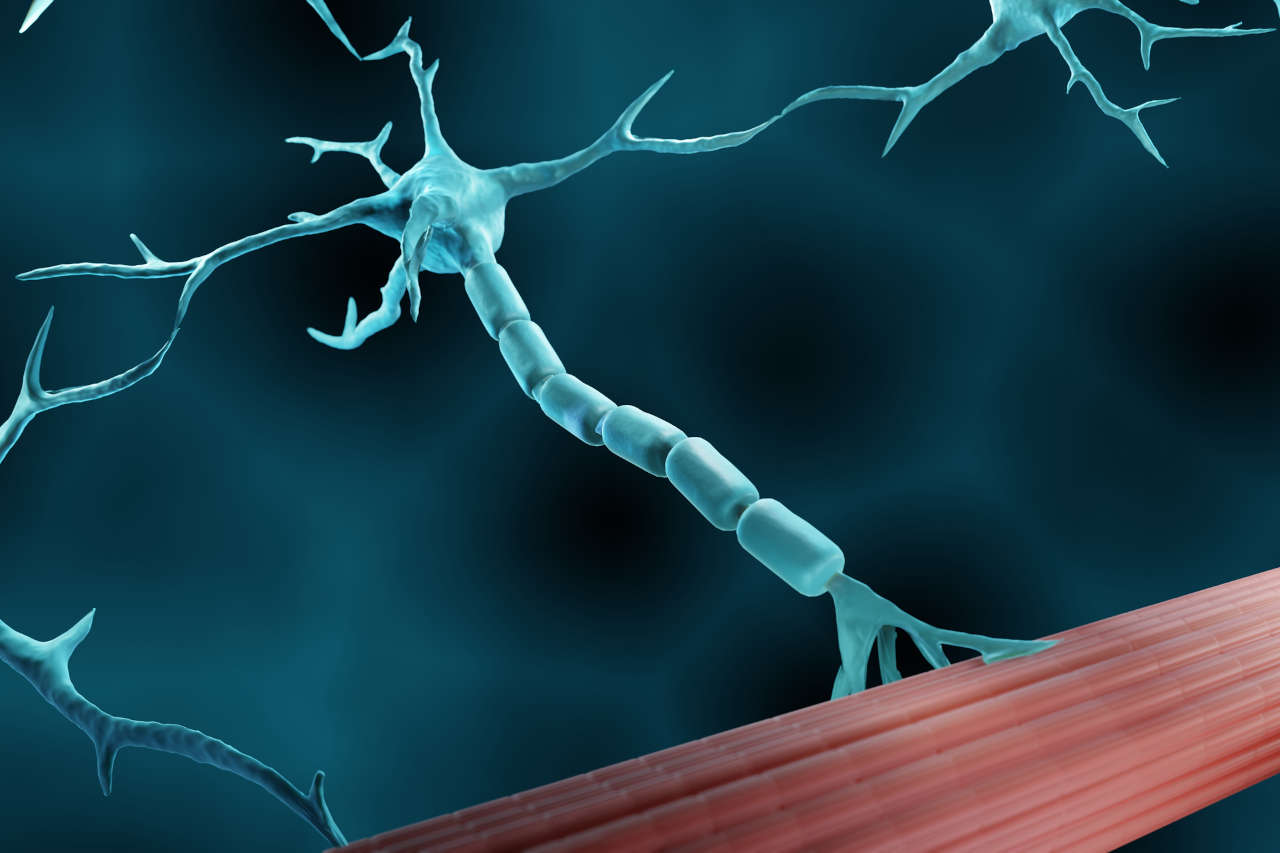
El síndrome de Lambert-Eaton (SMLE) es un trastorno poco común que se produce como resultado de un ataque del sistema inmunitario a la unión neuromuscular (el punto donde los nervios envían señales a los músculos). El daño en la unión neuromuscular provoca una transmisión deficiente de las señales nerviosas a los músculos, lo que provoca debilidad muscular gradual y una serie de otros síntomas.
Habla con un especialista
Acerca de la asistencia para copagosEsta condición a menudo se denomina síndrome miasténico de Lambert-Eaton debido a las similitudes de sus síntomas con miastenia gravis (un tipo de trastorno de la unión neuromuscular).
La prevalencia del síndrome de Lambert-Eaton es 46 veces menor que la de la miastenia gravis y afecta principalmente a hombres que a mujeres.
Por lo tanto, si a usted o a un ser querido le han diagnosticado recientemente LEMS, es importante que comprenda esta afección neurológica para poder controlarla adecuadamente.
Este artículo proporciona una comprensión básica del LEMS, incluida su causa subyacente, los signos y síntomas comunes y varias estrategias de tratamiento para ayudarlo a controlar esta afección.
Conceptos básicos del síndrome de Lambert-Eaton
Imagina que quieres realizar una actividad que requiere la función muscular. Tu sistema nervioso (cerebro) envía señales a través de los nervios a tus músculos para realizar esa acción. Estos nervios liberan una sustancia química llamada acetilcolina, que indica a las fibras musculares que se contraigan. La liberación de acetilcolina se facilita principalmente por los canales de calcio dependientes de voltaje presentes en los extremos de las terminaciones nerviosas. Por lo tanto, cuando estos canales se activan durante la transmisión de señales nerviosas, estimulan la liberación de acetilcolina, que se une a sus receptores en la fibra muscular y provoca la contracción muscular.
Sin embargo, en las personas con síndrome de Lambert-Eaton, el sistema inmunitario produce autoanticuerpos que bloquean los canales de calcio en las terminaciones nerviosas. Este bloqueo provoca una menor liberación de acetilcolina, insuficiente para la contracción muscular normal, y como resultado, las personas con LEMS experimentan debilidad muscular y fatiga.
La enfermedad recibe su nombre de Edward Lambert y Lee Eaton, dos neurólogos que describieron por primera vez los síntomas del síndrome miasténico en la década de 1950.
Causa subyacente del LEMS
Los investigadores creen que el sistema inmunitario es el factor determinante de la aparición del LEMS. Además, en el 50% al 60% de los casos, esta enfermedad también está altamente asociada. con ciertos tipos de cáncer, en particular el cáncer de pulmón de células pequeñas (CPCP).
Se teoriza que cuando el sistema inmunitario combate las células cancerosas, también ataca por error las terminaciones nerviosas, ya que estas contienen algunas de las mismas proteínas presentes en ellas. Esta confusión se denomina «síndrome paraneoplásico», que puede manifestarse como LEMS.
Sin embargo, no todos los cánceres causan LEMS. Actualmente se desconoce el desencadenante del LEMS sin cáncer; sin embargo, se teoriza que genes vinculados a la autoinmunidad podrían ser la causa. Las personas diagnosticadas con LEMS sin cáncer también son mucho más jóvenes, con una edad promedio de inicio de 35 años.
Signos y síntomas comunes del LEMS
La aparición de los síntomas del LEMS es gradual y suele desarrollarse en semanas o meses. Sin embargo, en pacientes con cáncer de pulmón de células pequeñas, la progresión de los síntomas del LEMS podría ser rápida.
El síntoma común del LEMS es la debilidad muscular proximal, y la distribución de la debilidad muscular comienza en las extremidades inferiores (parte superior de las piernas y caderas) y se desplaza hacia arriba hasta los músculos del hombro.

Las personas con LEMS pueden experimentar dificultad para caminar, subir escaleras y levantarse de sillas debido a la debilidad muscular de la cadera. Con el tiempo, la debilidad muscular mejora temporalmente tras la actividad muscular repetitiva, lo cual es una característica única de esta afección.
Otros síntomas del LEMS incluyen:
- Sensación de hormigueo en las manos o los pies.
- Rigidez y dolor muscular
- Fatiga
- Dificultad para caminar
- Debilidad oculobulbar, observada en aproximadamente el 701% de los pacientes. La debilidad ocular incluye ptosis y diplopía.
- Disfagia (dificultad para tragar)
- Disartria (dificultad para hablar)
- Insuficiencia respiratoria (poco frecuente, pero puede ocurrir en la etapa avanzada del LEMS)
La disfunción autonómica también es una preocupación frecuente en el LEMS y se observa en aproximadamente entre el 80 y el 96 % de los pacientes. Los síntomas más frecuentes son sequedad bucal, estreñimiento y disfunción eréctil.
¿Puede ayudar la IgIV?
Información gratuita sobre el tratamiento con IgIVDiagnóstico y pruebas para el LEMS
Los neurólogos realizan una combinación de pruebas, incluyendo una evaluación física. El neurólogo también considerará el historial médico y de medicación del paciente y observará los síntomas. Otras pruebas médicas incluyen:
Análisis de sangre
Esta prueba verifica la presencia de autoanticuerpos En el sistema circulatorio del paciente. Aproximadamente entre el 85% y el 95% de las personas con LEMS presentan estos autoanticuerpos en la sangre.
Electromiografía
Esta prueba verifica cómo están funcionando sus nervios y músculos.
Tomografía computarizada (TC), resonancia magnética (RM) o rayos X
Las tomografías computarizadas, las resonancias magnéticas y las radiografías se utilizan para comprobar neoplasias malignas subyacentes, como la presencia de cáncer de pulmón de células pequeñas (CPCP).
En algunos casos, el LEMS puede aparecer de 2 a 3 años antes que el cáncer, y si a una persona se le diagnostica LEMS, los proveedores de atención médica recomiendan exámenes de detección de cáncer cada 3 a 6 meses durante al menos 2 años.
Opciones de tratamiento para el síndrome de Lambert-Eaton
Aunque no existe una cura conocida para el LEMS, ciertos medicamentos y enfoques terapéuticos pueden ayudar a los pacientes a controlar sus síntomas. El objetivo principal del tratamiento es tratar la causa subyacente, como el cáncer, y controlar los síntomas del LEMS. Por lo tanto, si tiene LEMS asociado a un cáncer como el CPCP, tratar primero la causa subyacente (el cáncer) puede ayudar a tratar el LEMS.
Los proveedores de atención médica recomiendan una combinación de cirugía, quimioterapia y radioterapia según el tipo de cáncer que tenga el paciente.
En el caso del tratamiento del LEMS, generalmente se utilizan medicamentos diseñados para reducir los síntomas del LEMS y terapias inmunomoduladoras que suprimen la respuesta inmunitaria/ataque inmunológico a los nervios.
Los medicamentos más eficaces que se recomiendan como tratamiento de primera línea para reducir los síntomas del LEMS incluyen:
- Amifampridina (Firdapse):Este medicamento aumenta la liberación de acetilcolina y, por lo tanto, mejora la transmisión de señales a los músculos. La dosis inicial recomendada de Firdapse para adultos es de 15 a 30 mg al día, dividida en tres o cuatro dosis. Su profesional de la salud puede aumentar la dosis hasta un máximo de 80 mg al día.
- Guanidina: Este medicamento también aumenta la liberación de acetilcolina y mejora las contracciones musculares. La dosis inicial recomendada de guanidina es de 15 a 30 mg/kg/día, dividida en tres o cuatro dosis. La dosis puede aumentarse según la tolerabilidad hasta un máximo de 35 mg/kg/día.
- Piridostigmina (Mestinón): Este medicamento actúa previniendo la degradación de la acetilcolina en la unión entre los nervios y los músculos. Al aumentar la cantidad de acetilcolina disponible, puede ayudar a aumentar la fuerza muscular. El uso de Mestinon para esta indicación se considera fuera de indicación, lo que significa que no está aprobado por la FDA para este uso. La dosis de Mestinon para el LEMS varía, ya que el rango es amplio y se basa en la tolerabilidad. Su profesional de la salud le recetará la dosis adecuada.
A veces, cuando la medicación no funciona bien y el paciente persiste con una debilidad persistente, los profesionales de la salud recomiendan terapias inmunomoduladoras o inmunosupresoras que suprimen la respuesta inmunitaria hiperactiva. Estas terapias incluyen:
- Terapia con inmunoglobulina (IVIG), un procedimiento en el que se inyectan anticuerpos en el cuerpo que impiden temporalmente que el sistema inmunitario ataque los nervios.
- Plasmaféresis, un procedimiento para filtrar los autoanticuerpos del cuerpo, previniendo así el daño a los nervios.
- Medicamentos inmunosupresores para reducir la actividad de su sistema inmunológico.
Además de los medicamentos y las terapias inmunosupresoras, un estilo de vida saludable puede ayudar a prevenir la aparición del LEMS. Dado que, en el 50% de los casos, la aparición del LEMS se asocia con cáncer de pulmón, evitar el tabaco puede ayudar a prevenir el cáncer.
Pronóstico del síndrome de Lambert-Eaton
Siempre que la afección subyacente se trate con prontitud, el pronóstico del LEMS es favorable. Los tratamientos y fármacos inmunosupresores generalmente tuvieron un efecto positivo en los pacientes con LEMS, aumentando sus probabilidades de supervivencia y mejorando su calidad de vida.
Después del diagnóstico, 85% de los pacientes recuperan la independencia en las actividades diarias dentro de un año de tratamiento.
REFERENCIAS:
- Pascuzzi, RM, y Bodkin, CL (2022). Miastenia grave y síndrome miasténico de Lambert-Eaton: nuevos avances en diagnóstico y tratamiento. Enfermedades neuropsiquiátricas y su tratamiento, 18, 3001-3022. https://doi.org/10.2147/NDT.S296714
- Kesner, VG, Oh, SJ, Dimachkie, MM y Barohn, RJ (2018). Síndrome miasténico de Lambert-Eaton. Neurologic Clinics, 36(2), 379-394. https://doi.org/10.1016/j.ncl.2018.01.008
- Titulaer, MJ, Lang, B. y Verschuuren, JJ (2011). Síndrome miasténico de Lambert-Eaton: de las características clínicas a las estrategias terapéuticas. The Lancet Neurology, 10(12), 1098-1107. https://doi.org/10.1016/S1474-4422(11)70245-9
- Sanders, DB (2003). Síndrome miasténico de Lambert-Eaton. Anales de la Academia de Ciencias de Nueva York, 998(1), 500-508. https://doi.org/10.1196/annals.1254.065
- Harper, CM, y Lennon, VA (2018). Síndrome de Lambert-Eaton. Miastenia grave y trastornos relacionados, 221-237. https://doi.org/10.1007/978-3-319-73585-6_14
- Merino-Ramírez, M. Á., y Bolton, CF (2016). Revisión de los desafíos diagnósticos del síndrome de Lambert-Eaton revelados a través de tres informes de casos. Revista Canadiense de Ciencias Neurológicas, 43(5), 635-647.
- Hülsbrink, R., y Hashemolhosseini, S. (2014). Síndrome miasténico de Lambert-Eaton: diagnóstico, patogénesis y tratamiento. Neurofisiología clínica, 125(12), 2328-2336. https://doi.org/10.1016/j.clinph.2014.06.031
- Ivanovski, T. y Miralles, F. (2019). Síndrome miasténico de Lambert-Eaton: el diagnóstico temprano es clave. Enfermedades neurológicas y neuromusculares degenerativas, 27-37. https://doi.org/10.2147/DNND.S192588
- Gilhus, NE (2011). Síndrome miasténico de Lambert-Eaton: patogénesis, diagnóstico y tratamiento. Enfermedades autoinmunes, 2011. https://doi.org/10.4061/2011/973808
- McEvoy, KM, Windebank, AJ, Daube, JR y Low, PA (1989). 3,4-Diaminopiridina en el tratamiento del síndrome miasténico de Lambert-Eaton. The New England Journal of Medicine, 321(23), 1567-1571. https://doi.org/10.1056/nejm198912073212303